

Department of anatomy and Physiology and Lipid Research Unit, Centre Hospitalier Universitaire de l'Université Laval (CHUL) Research Centre, Quebec City, Quebec, Canada
andre.marette@crchul.ulaval.ca

Insulin resistance is defined as reduced insulin action in metabolic and vascular target tissues. Whereas it is widely recognized that insulin resistance is a key pathogenic factor in the development of diabetes and cardiovascular disease (CVD), its etiology remains elusive. In this short report, we will summarize our research efforts toward establishing the potential mechanisms responsible for promoting insulin resistance in key metabolic tissues.
Inflammation as a cause of insulin resistanceWhile obesity-linked diabetes and CVD are known to be chronic inflammatory disorders, the underlying mechanisms by which inflammation promotes these metabolic diseases remain poorly understood. Studies in my laboratory identified inducible nitric oxide synthase (iNOS) as a key inflammatory mediator in obesity, causing insulin resistance in skeletal muscle [1-4] (Figure 1) and impairing insulin action in the liver through inhibition of adiponectin secretion by adipose tissue [5] (Figure 1). Studies by other groups have confirmed the role of iNOS in obesity-linked insulin resistance [6-8] and further indicated that iNOS induction in blood vessels is also involved in mediating vascular dysfunction in obesity [8]. The underlying cause of inflammation in obesity remains poorly understood, but one theory is that it lies within the origin of fat cells. Indeed, metabolic and immune pathways have evolved to be closely linked and interdependent. The finding that obesity is characterized by macrophage accumulation in adipose tissue [9, 10] and that macrophages and fat cells share the expression of multiple genes has added another dimension to our understanding of the development of adipose tissue inflammation in obesity. The role of immune cells in promoting inflammation in obesity has also recently been confirmed in humans [11-13]. What remains to be determined is how obesity promotes an inflammatory process not only in adipose tissue, but also in skeletal/cardiac muscles and liver. In this regard, recent studies point toward hypoxia as a key triggering event in the development of an inflammatory state in obesity [14-16]. However, this remains to be confirmed and better characterized, especially in human obesity.
Nutrient sensing through the mTOR pathway promotes insulin resistance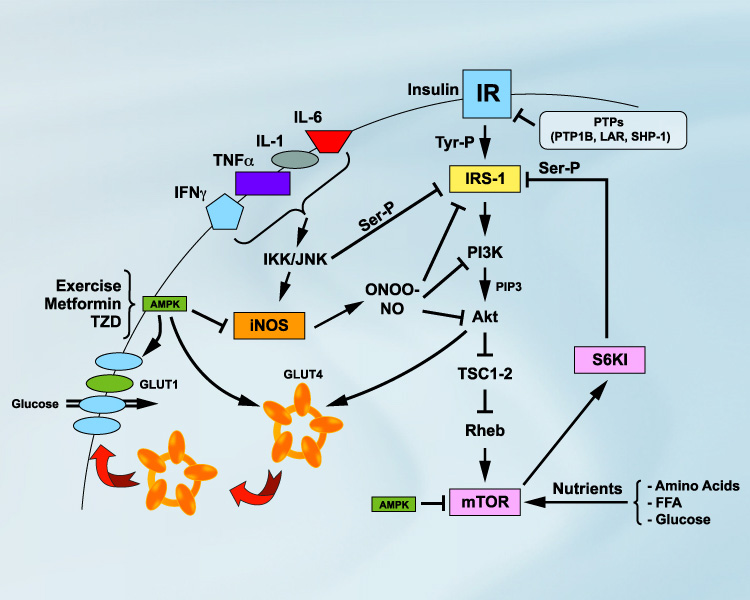 [Click to enlarge]
[Click to enlarge]
Detailed legend of Figure 1 Proinflammatory cytokines (TNFα, IL-1, IL-6 and IFNγ) are released from adipocytes or infiltrating macrophages in adipose tissue or skeletal muscle. This leads to cytokine signalling events, including activation of c-jun N-terminal kinase (JNK) and IkB kinase (IKK). IKK and JNK can promote insulin resistance by increasing inhibitory serine phosphorylation of IRS-1, a key element of the insulin signalling cascade, or through the transcriptional activation of inflammatory genes such as iNOS. iNOS activation leads to high levels of nitric oxide (NO) production and formation of the highly reactive derivative peroxynitrite (ONOO-). NO and ONOO- are thought to impede insulin signalling by s-nitrosylation or nitration of IRS-1, PI3K and/or Akt, which are key to glucose transporter 4 (GLUT4) translocation to the cell surface and activation of glucose transport in the myocytes. Prolonged hyperinsulinemia and nutrient satiation also activates the mTOR/S6K1 pathway, causing insulin resistance by enhancing phosphorylation of IRS-1 on multiple serine residues. Conversely, activation of AMP-activated protein kinase (AMPK) by physical exercise or pharmacological means (TZD and metformin) improves insulin action through inhibition of iNOS as well as mTOR/S6K1 signalling. AMPK can also increase glucose transport by triggering GLUT4 translocation and activating the cell surface glucose transporters. The protein tyrosine phosphatases (PTPs) PTP1B, LAR and SHP-1 may also mediate insulin resistance by dephosphorylation of key tyrosine residues within the insulin receptor
Download PDF version of this figureRecent studies by our group and others suggest that nutrient satiation promotes insulin resistance by activating the protein kinase mTOR pathway, a sensing complex that integrates nutrient and hormonal signals [17-21]. We first proposed that mTOR operates a negative feedback loop by phosphorylating the first substrate of the insulin receptor, IRS-1, on multiple serine residues, uncoupling IRS-1 from the activation of phosphatidylinositol 3-kinase (PI3K) and Akt, two effectors of insulin's metabolic actions [20] (Figure 1). This metabolic feedback loop has been found in myocytes [20, 22], adipocytes and hepatocytes [17, 23] as well as in liver and muscle tissues of rats [17], suggesting that the mTOR pathway plays a major role in the regulation of glucose homeostasis. Importantly, we and others have shown that mTOR and its effector S6K1 are "overactivated" in skeletal muscle, liver and adipose tissue of both genetic and dietary animal models of obesity-linked insulin resistance [17, 24]. We have further shown that the mTOR pathway negatively modulates insulin's metabolic actions in skeletal muscle and adipocytes of healthy subjects [17, 22]. We have also recently identified that serine 1101 in the IRS-1 protein is a molecular target of S6K1 in the liver of obese animals and in skeletal muscle during infusion of human subjects with amino acids [25]. Whether increased activation of S6K1 is a common feature of human obesity and insulin resistance is currently unknown, but IRS-1 (Ser-1101) and S6K1 (Thr-389) may represent future diagnostic tools in order to predict and design therapeutic treatments.
AMPK: turning on metabolism while turning off insulin resistanceAMPK is a member of a metabolite-sensing protein kinase family that acts as a fuel gauge monitoring cellular energy levels [26, 27]. When AMP kinase "senses" decreased energy stores, it acts to switch off ATP-consuming pathways and switch on alternative pathways for ATP regeneration. AMPK is activated by exercise/muscle contraction [28] but also by several classes of drugs that are currently used for treatment of diabetes and CVD, including thiazolidinedione (TZD), and that activate proliferator-activated receptor gamma (PPARg). We have recently reported that PPARg agonists inhibit iNOS induction in macrophages, myocytes and adipocytes through activation of AMPK [29] (Figure 1). These studies indicate that AMPK is a master switch that turns on metabolic pathways while turning off inflammation in insulin target tissues and macrophages. Interestingly, AMPK may also improve insulin sensitivity by blunting the activation of the mTOR/S6K1 pathway (Figure 1). Indeed, activation of AMPK by the pharmacological activator AICAR or by the anti-diabetic drug metformin inhibits mTOR/S6K1 in various cell types [30, 31]. AMPK may therefore represent a key therapeutic target since its activation can blunt both inflammation and nutrient sensing signals believed to play a key role in promoting insulin resistance in obesity.
SHP-1: a new target for the treatment of insulin resistance?Because tyrosine phosphorylation is key to insulin signal transduction, protein tyrosine phosphatases (PTPs) are prominent candidates to negatively regulate insulin action. Previous studies have shown that the PTPs PTP1B and LAR (leukocyte related-antigen) are negative regulators of the insulin receptor kinase in liver and peripheral insulin target tissues [32-34]. PTP1B-deficient mice are leaner, exhibit increased energy expenditure and are protected from insulin resistance in the liver and skeletal muscle [35, 36]. Neuron-specific PTP1B KO also increased leptin sensitivity and improved glucose homeostasis, suggesting that PTP1B regulates body mass and adiposity primarily through actions in the brain [37].
We have recently identified the PTP SHP-1 as a novel inhibitor of insulin receptor signalling in liver and skeletal muscle [38]. We found that mouse models with a functionally deficient SHP-1 protein are remarkably glucose tolerant and insulin sensitive for glucose metabolism as a result of increased insulin signalling to the IRS/PI3K/Akt pathway in both liver and muscle tissues. These findings indicate that SHP-1 plays an important role in the regulation of insulin signalling in liver and muscle. Preliminary data also demonstrates that SHP-1 is expressed in adipose tissue and modulates lipid metabolism and adiposity (A. Marette, unpublished data) but the mechanisms involved remain poorly understood. It will be important in the near future to clarify the role of SHP-1 in controlling insulin sensitivity in insulin-resistant states and investigate whether this PTP is a potential target for anti-diabetic drugs.
Concluding remarksGiven the prevalence of obesity worldwide and the increase in associated health complications such as diabetes and CVD, the need for a mechanistic understanding of obesity-related insulin resistance remains a major research priority. We also need to speed up the discovery of new biological markers and diagnosis tools to assess insulin resistance and predict its development in populations at risk. Finally, it is critical to find novel therapeutic targets to improve the pharmacotherapy of obese diabetic subjects, which is a crucial measure when lifestyle modifications (e.g., physical activity, diets) fail to achieve the therapeutic goals.
References
- Bédard S, Marcotte B and Marette A. Cytokines modulate glucose transport in skeletal muscle by inducing the expression of inducible nitric oxide synthase. Biochem J 1997; 325 (Pt 2): 487-93.
- Kapur S, Bedard S, Marcotte B, et al. Expression of nitric oxide synthase in skeletal muscle: a novel role for nitric oxide as a modulator of insulin action. Diabetes 1997; 46: 1691-700.
- Marette A. Mediators of cytokine-induced insulin resistance in obesity and other inflammatory settings. Curr Opin Clin Nutr Metab Care 2002; 5: 377-83.
- Perreault M and Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med 2001; 7: 1138-43.
- Dallaire P, Laplante P, Penfornis P, et al. Obese mice lacking iNOS are sensitised to the metabolic actions of PPARg agonism. Diabetes 2007 (in revision).
- Carvalho-Filho MA, Ueno M, Hirabara SM, et al. S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes 2005; 54: 959-67.
- Fujimoto M, Shimizu N, Kunii K, et al. A role for iNOS in fasting hyperglycemia and impaired insulin signaling in the liver of obese diabetic mice. Diabetes 2005; 54: 1340-8.
- Noronha BT, Li JM, Wheatcroft SB, et al. Inducible nitric oxide synthase has divergent effects on vascular and metabolic function in obesity. Diabetes 2005; 54: 1082-9.
- Weisberg SP, McCann D, Desai M, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003; 112: 1796-808.
- Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003; 112: 1821-30.
- Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res 2005; 46: 2347-55.
- Greenberg AS and Obin MS. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr 2006; 83: 461S-5S.
- Wu H, Ghosh S, Perrard XD, et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 2007; 115: 1029-38.
- Cancello R, Henegar C, Viguerie N, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 2005; 54: 2277-86.
- Segawa K, Fukuhara A, Hosogai N, et al. Visfatin in adipocytes is upregulated by hypoxia through HIF1alpha-dependent mechanism. Biochem Biophys Res Commun 2006; 349: 875-82.
- Semeraro F, Voros G, Collen D, et al. Impairment of adipose tissue development by hypoxia is not mediated by plasminogen activator inhibitor-1. Thromb Haemost 2006; 95: 174-81.
- Khamzina L, Veilleux A, Bergeron S, et al. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 2005; 146: 1473-81.
- Korsheninnikova E, van der Zon GC, Voshol PJ, et al. Sustained activation of the mammalian target of rapamycin nutrient sensing pathway is associated with hepatic insulin resistance, but not with steatosis, in mice. Diabetologia 2006; 49: 3049-57.
- Tremblay F, Jacques H and Marette A. Modulation of insulin action by dietary proteins and amino acids: role of the mammalian target of rapamycin nutrient sensing pathway. Curr Opin Clin Nutr Metab Care 2005; 8: 457-62.
- Tremblay F and Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem 2001; 276: 38052-60.
- Um SH, D'Alessio D and Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab 2006; 3: 393-402.
- Tremblay F, Krebs M, Dombrowski L, et al. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes 2005; 54: 2674-84.
- Tremblay F, Gagnon A, Veilleux A, et al. Activation of the mammalian target of rapamycin pathway acutely inhibits insulin signaling to Akt and glucose transport in 3T3-L1 and human adipocytes. Endocrinology 2005; 146: 1328-37.
- Um SH, Frigerio F, Watanabe M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004; 431: 200-5.
- Tremblay F, Brule S, Hee Um S, et al. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci U S A 2007; 104: 14056-61.
- Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem Sci 2004; 29: 18-24.
- Hardie DG. AMP-activated protein kinase: a key system mediating metabolic responses to exercise. Med Sci Sports Exerc 2004; 36: 28-34.
- Lemieux K, Han XX, Dombrowski L, et al. The transferrin receptor defines two distinct contraction-responsive GLUT4 vesicle populations in skeletal muscle. Diabetes 2000; 49: 183-9.
- Pilon G, Dallaire P and Marette A. Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. J Biol Chem 2004; 279: 20767-74.
- Ju JS, Gitcho MA, Casmaer CA, et al. Potentiation of insulin-stimulated glucose transport by the AMP-activated protein kinase. Am J Physiol Cell Physiol 2007; 292: C564-72.
- Tzatsos A and Kandror KV. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol 2006; 26: 63-76.
- Asante-Appiah E and Kennedy BP. Protein tyrosine phosphatases: the quest for negative regulators of insulin action. Am J Physiol Endocrinol Metab 2003; 284: E663-70.
- Elchebly M, Cheng A and Tremblay ML. Modulation of insulin signaling by protein tyrosine phosphatases. J Mol Med 2000; 78: 473-82.
- Goldstein BJ. Protein-tyrosine phosphatases: emerging targets for therapeutic intervention in type 2 diabetes and related states of insulin resistance. J Clin Endocrinol Metab 2002; 87: 2474-80.
- Elchebly M, Payette P, Michaliszyn E, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999; 283: 1544-8.
- Klaman LD, Boss O, Peroni OD, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol 2000; 20: 5479-89.
- Bence KK, Delibegovic M, Xue B, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med 2006; 12: 917-24.
- Dubois MJ, Bergeron S, Kim HJ, et al. The SHP-1 protein tyrosine phosphatase negatively modulates glucose homeostasis. Nat Med 2006; 12: 549-56.
