

ulf.smith@medic.gu.se

Visceral (intra-abdominal) fat has attracted much interest both as a marker and an inducer of many of the metabolic abnormalities associated with obesity [1]. It is now well established that abdomi-nal circumference, which is used as a surrogate marker of visceral fat, is an easily available clinical measurement of both cardiovascular risk and type 2 diabetes risk in obesity. Consequently, it has become a key measurement for the definition of the metabolic syndrome [2].
We know that an expanded visceral fat mass, when measured as waist circumference, is a better predictor of insulin resistance, dyslipidemia, low-grade inflammation, and other risk factors for both type 2 diabetes and cardiovascular disease than obesity measured with body mass index. However, the key question is whether, or to what extent, increased visceral fat leads to these risk factors or if it is mainly a marker.
We know that visceral adipose cells have an increased lipolytic rate and that they respond poorly to the antilipolytic effect of insulin. This has led to the wide-spread hypothesis that elevated free fatty acid (FFA) levels in the portal blood increase hepatic VLDL triglyceride secretion as well as induce liver steatosis and hepatic insulin resistance with an accompanying enhanced hepatic glu-cose production. In addition, visceral adipose tissue has been shown to have an increased expres-sion of different inflammatory proteins and cytokines [1]. It was also recently shown that visceral fat has an increased interleukin (IL)-6 release [3] which, in turn, increases C-reactive protein (CRP) release by the liver. CRP is a key marker of inflammation and a well-established risk marker of both cardiovascular disease and type 2 diabetes.
Although it has now been well documented in rodents, the "hyper-lipolytic" effect of visceral fat in man is conjectural. in vivo studies in man have clearly shown that the visceral depot only makes a minor contribution to circulating FFA levels. Furthermore, there is no convincing evidence in man that portal FFA levels are higher than systemic levels.
So if this is true, why is amount of visceral fat so clearly associated with the metabolic syndrome? In my opinion, this is because visceral fat is predominantly a marker of ectopic fat and, thus, is like several other ectopic sites that expand when the subcutaneous depot is unable to store the excess calories we so easily consume in abundance. This does not at all detract from the importance of visceral fat as an important clinical marker of metabolic cardiovascular risk. In addition, visceral fat also contributes to the proinflammatory state of obesity and the induction of CRP in the liver. A correlation between visceral fat, liver fat, and VLDL triglyceride synthesis has also been shown [4]. However, this may not be a direct and causal effect but rather reflect increased ectopic lipid storage in many sites: in adipose cells in other depots, in skeletal and cardiac muscle cells, as well as in the liver. In addition, the fact that the portal vein drains the visceral fat makes the liver an important target for the released adipokines (Figure). IL-6 seems particularly abundant in the visceral fat [3]. Chronically elevated IL-6 induces insulin resistance in both the liver and the adi-pose cells.
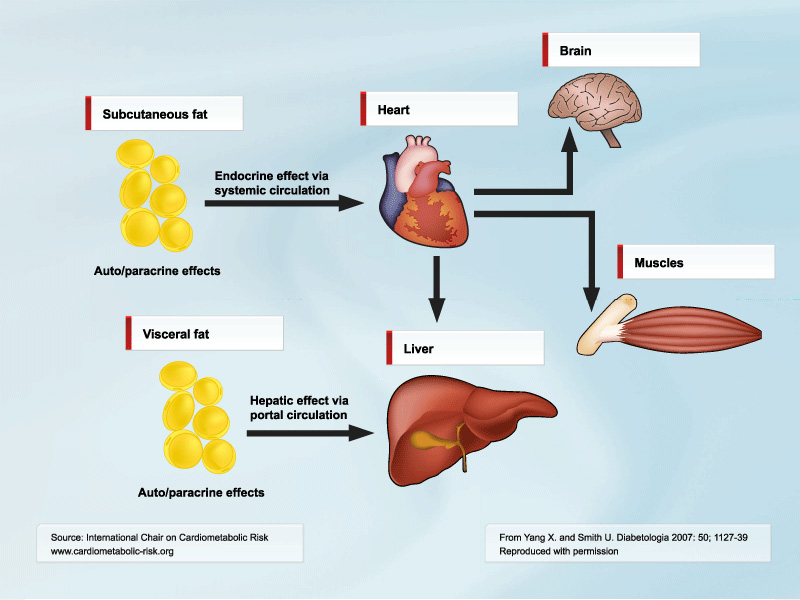 [Click to enlarge]
[Click to enlarge]
Legend: The cytokines released from visceral depot would primarily alter carbohydrate and lipid metabolism and stimulate production of acute-phase response proteins in the liver, whereas the cytokines produced by subcutaneous depot would mainly affect adipose cell development and function locally as well as exert systemic effects on, e.g., the skeletal muscle.
Download PDF version of this figureIf we want to identify the basic problem, we should probably look at the ability of subcutaneous tissue to form new adipose cells and how that process is regulated. Interestingly, the subcutaneous cells are inappropriately large in non-obese but insulin-resistant individuals. We have also found a negative correlation between average fat cell size of the abdominal subcutaneous adipose tissue and insulin sensitivity measured with the euglycemic clamp technique in non-obese and healthy first-degree relatives of type 2 diabetic subjects. The inappropriately enlarged fat cells may well be an indication of an inability to recruit new preadipocytes to store the lipids in the subcutaneous tissue. In fact, several animal models have shown that insulin resistance and other metabolic consequences of obesity are prevented if the animals are genetically engineered to allow abundant recruitment of new preadipocytes in the subcutaneous tissue [5].
It is not the purpose of this article to review the complicated steps of adipogenesis but merely to emphasize the role of several ectopic fat depots, including the visceral depot, as "spill-over" sites for lipids when the subcutaneous sites become insufficient. However, these ectopic sites also con-sist of adipose cells producing different adipokines, including cytokines and chemokines, which may exert local effects (e.g., endothelial dysfunction if located around the vessels) as well as con-tribute to overall systemic release.
Corroborating evidence for the view that visceral fat is an ectopic fat depot comes from recent studies where epicardial fat has been measured. Amount of epicardial fat correlates with amount of visceral fat [4]. The negative metabolic associations are, as with visceral fat, also found for amount of epicardial fat, and this holds true for inflammatory markers [6, 7].
Thus, we are now entering a phase where we should examine the subcutaneous adipose tissue and its ability to recruit and develop new adipose cells in obesity. The role of genetic factors is un-known. It is also unclear how the mesenchymal stem cells become committed to preadipocytes and subsequently develop into mature adipose cells. Furthermore, induction of inflammation in the subcutaneous adipose tissue in obesity impairs the development of preadipocytes to adipocytes [8]. Understanding the molecular mechanisms for adipogenesis in man is likely to open new avenues to the prevention of obesity-associated complications, in part by preventing the accumulation of ectopic fat depots such as visceral fat.
References
- Yang X and Smith U. Adipose tissue distribution and risk of metabolic disease: does thiazolidinedione-induced adi-pose tissue redistribution provide a clue to the answer? Diabetologia 2007; 50: 1127-39.
- The IDF consensus worldwide definition of the metabolic syndrome. http://www.idf.org/webdata/docs/Metac_syndrome_def.pdf. 2005; 2006.
- Fontana L, Eagon JC, Trujillo ME, et al. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007; 56: 1010-3.
- Adiels M, Westerbacka J, Soro-Paavonen A, et al. Acute suppression of VLDL1 secretion rate by insulin is associated with hepatic fat content and insulin resistance. Diabetologia 2007; 50: 2356-65.
- Kim JY, van de Wall E, Laplante M, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 2007; 117: 2621-37.
- Iacobellis G and Leonetti F. Epicardial adipose tissue and insulin resistance in obese subjects. J Clin Endocrinol Metab 2005; 90: 6300-2.
- Cikim AS, Topal E, Harputluoglu M, et al. Epicardial adipose tissue, hepatic steatosis and obesity. J Endocrinol Invest 2007; 30: 459-64.
- Gustafson B and Smith U. Cytokines promote Wnt signaling and inflammation and impair the normal differentiation and lipid accumulation in 3T3-L1 preadipocytes. J Biol Chem 2006; 281: 9507-16.
